
La distonía es una enfermedad muy heterogénea, ya que puede darse por causas adquiridas o por fallos genéticos. Dentro de las causas genéticas, se han descrito hasta ahora más de 200 genes que causan distonía.
El diagnóstico genético se define como el análisis del ADN para detectar mutaciones o cambios hereditarios con un objetivo clínico. En el caso de la distonía, al igual que en otras enfermedades del sistema nervioso, su diagnóstico temprano disminuye la necesidad de realizar más pruebas diagnósticas, permite conocer el pronóstico y asesorar a las familias, por lo que también tiene un impacto psicosocial muy positivo para el paciente y la familia.
A continuación, exponemos una serie de conceptos clave que nos ayudan a entender la genética y el diagnóstico de la distonía:
Nuestro ADN es una gran biblioteca que contiene toda la información que condiciona el funcionamiento de nuestro cuerpo. Cada uno de los libros que forman esta biblioteca son nuestros genes, y un error en alguno de ellos (una falta de ortografía, la falta de alguna palabra…) es lo que llamamos una mutación. Las mutaciones hacen que los genes no se puedan leer correctamente, lo que altera la fabricación de proteínas y, por ende, algunas funciones de nuestro cuerpo, dando lugar a diferentes enfermedades.
Una de las maneras que tiene nuestro cuerpo de protegernos frente a las enfermedades genéticas es que nuestro ADN está duplicado. Todos tenemos dos copias de cada gen, una que heredamos de nuestro padre y otra de nuestra madre. De este modo, la aparición de las enfermedades genéticas está condicionada al número de copias que tenemos alteradas (mutadas):
-
-
Expresión dominante. Este patrón se produce cuando una mutación en una única copia de un gen es suficiente para expresar una enfermedad.
-

-
Expresión recesiva. En este caso, se requiere que ambas copias de un gen estén alteradas para que se produzca la enfermedad, es decir, la copia de la madre y del padre tienen la mutación.

-
Expresión ligada al cromosoma X. El cromosoma X también se denomina cromosoma sexual porque determina el sexo de los seres vivos. Así, las personas de sexo femenino tienen dos cromosomas X y las de sexo masculino solo tienen uno, quedando más vulnerables a las mutaciones en los genes de este cromosoma, ya que solo tienen una copia. Al igual que ocurre con los genes localizados en los demás cromosomas, las enfermedades causadas por mutaciones en el cromosoma X pueden seguir un patrón dominante, en el cual tanto hombres como mujeres presentarán la enfermedad, o recesivo, en el que las mujeres pueden o no presentar la enfermedad.


-
Expresión mitocondrial. Ocurre cuando existen alteraciones en una pequeña proporción del ADN que se encuentra en las mitocondrias de la célula (ADN mitocondrial). Se trata de un patrón característico ya que las mitocondrias se heredan únicamente de la madre, por lo que sólo las mujeres pueden transmitir la mutación.

Algunas veces, las mutaciones pueden heredarse a partir de alguna copia alterada de los padres. Por el contrario, hay ocasiones en las que las mutaciones se producen de manera espontánea. Estas mutaciones, llamadas de novo, son aquellas que aparecen por primera vez en el niño afecto, y está ausente en el ADN de ambos padres. Sin embargo, esta mutación se podrá ir transmitiendo si el niño afecto tiene descendencia en el futuro. De hecho, todas las mutaciones heredadas de los padres fueron en algún momento de novo y aparecieron espontáneamente en un antepasado de la familia.

Por último, además de los principales tipos de herencia, hay algunos casos que no siguen estas reglas básicas. Esto se debe a que existen otros factores menos conocidos que hacen que cada persona manifieste la enfermedad de maneras distintas:
-
Se define como la proporción de individuos con una mutación que presentan la enfermedad. Así, existen genes que presentan ‘penetrancia incompleta’, es decir, no todos los individuos manifiestan los síntomas de la enfermedad. Algunos genes que dan lugar a formas de distonía de penetrancia incompleta son el KMT2B o el TOR1A.
-
Este fenómeno se produce por una mutación que aparece muy temprano en el desarrollo del feto y hace que no todas las células del cuerpo contengan la mutación. Así, la expresión de la enfermedad será variable en función del tipo de células afectadas. Algunos genes que causan distonía y dan lugar a mosaicos son el GNAO1 o el ADCY5.
-
Impronta genética o imprinting. Es un fenómeno genético por el que la expresión de ciertos genes está condicionada por el sexo del progenitor del cual se heredó. Por ejemplo, en el caso de la distonía mioclónica (DYT11-SGCE), la copia del gen heredada de la madre está silenciada. De esta manera, una persona que tenga una mutación en la copia materna del gen no manifestará síntomas de la enfermedad, aunque será portadora de ella y podrá transmitirla a su descendencia.
-
Distonías de herencia recesiva: Un claro ejemplo son muchas de las distonías causadas por errores congénitos del metabolismo.
-
Distonías dominantes hereditarias: A este grupo pertenecen la mayoría de distonías aisladas, que están clasificadas según la nomenclatura DYT (Ver tabla abajo). Un ejemplo de esta categoría es la distonía mioclónica (DYT-SGCE), ya que el silenciamiento de la enfermedad causada por mutaciones maternas hace que más del 80% de los casos sean heredados del padre.
-
Distonías dominantes de novo: Son formas graves de la enfermedad, como las causadas por mutaciones en el gen GNAO1, por lo que su transmisión a la descendencia es difícil.
-
Distonías de herencia mitocondrial: Son formas progresivas de distonía que pueden tener grados de severidad muy variables. Los niños con este tipo de distonía causada por mutaciones en el ADN mitocondrial sufren una enfermedad que se denomina síndrome de Leigh. Este síndrome ocurre cuando, debido a un defecto en el metabolismo mitocondrial, el cerebro no puede generar la energía suficiente para funcionar correctamente, produciéndose lesiones en el cerebro, más concretamente en los ganglios de la base.
-
Distonías ligadas al cromosoma X: Algunos ejemplos de distonías ligadas a mutaciones en el cromosoma X son la distonía-parkinsonismo (DYT3) o el síndrome de sordera-distonía ligado al cromosoma X.
|
|
Tipo de distonía |
Herencia |
Gen |
|
DYT1 |
Distonia generalizada de inicio temprano |
Dominante |
TOR1A |
|
DYT2 |
Distonía idiopática autosómica recesiva |
Recesiva |
HPCA |
|
DYT3 |
Distonía con parkinsonismo ligado al X |
Recesiva ligada a X |
TAF1 |
|
DYT4 |
Distonía con disfonía en susurro |
Dominante |
TUBB4A |
|
DYT5 |
Distonía con respuesta a levodopa o distonía de Segawa |
Dominante/Recesiva |
GCH1 |
|
DYT6 |
Distonía mixta de inicio en la adolescencia |
Dominante |
THAP1 |
|
DYT7 |
Distonía focal de inicio en el adulto |
Dominante |
Desconocido |
|
DYT8 |
Disquinesia paroxística no quinesiogénica (PNKD1) |
Dominante |
PNKD |
|
DYT9 |
Coreoatetosis distónica paroxística con ataxia episódica |
Dominante |
SLC2A1 |
|
DYT10 |
Disquinesia paroxística quinesiogénica (PKD) |
Dominante |
PRRT2 |
|
DYT11 |
Distonía mioclónica (DYT-SGCE) |
Dominante |
SGCE |
|
DYT12 |
Distonía con parkinsonismo de inicio rápido |
Dominante |
ATP1A3 |
|
DYT13 |
Distonía primaria craneocervical de inicio temprano |
Dominante |
Desconocido |
|
DYT15 |
Distonía mioclónica |
Dominante |
Desconocido |
|
DYT16 |
Distonía con parkinsonismo de inicio en la adolescencia |
Recesiva |
PRKRA |
|
DYT17 |
Distonía primaria idiopática autosómica recesiva |
Recesiva |
Desconocido |
|
DYT18 |
Discinesia paroxística inducida por esfuerzo |
Dominante |
SLC2A1 |
|
DYT19 |
Disquinesia paroxística quinesiogénica 2 (EKD2) |
Dominante |
Desconocido |
|
DYT20 |
Disquinesia paroxística no quinesiogénica 2 (PNKD2) |
Dominante |
Desconocido |
|
DYT21 |
Distonía focal de inicio tardío |
Dominante |
Desconocido |
|
DYT23 |
Distonía cervical primaria de inicio en el adulto |
Dominante |
Desconocido |
|
DYT24 |
Distonía craneocervical autosómica dominante |
Dominante |
ANO3 |
|
DYT25 |
Distonía focal primaria autosómica dominante de inicio tardío |
Dominante |
GNAL |
|
DYT26 |
Distonía mioclónica |
Dominante |
KCTD17 |
|
DYT27 |
Distonía aislada de inicio temprano |
Recesiva |
COL6A3 |
|
DYT28 |
Distonía generalizada de inicio temprano |
Dominante |
KMT2B |
|
DYT29 |
Distonía de inicio en la infancia con atrofia óptica y anormalidad de los ganglios basales |
Recesiva |
MECR |
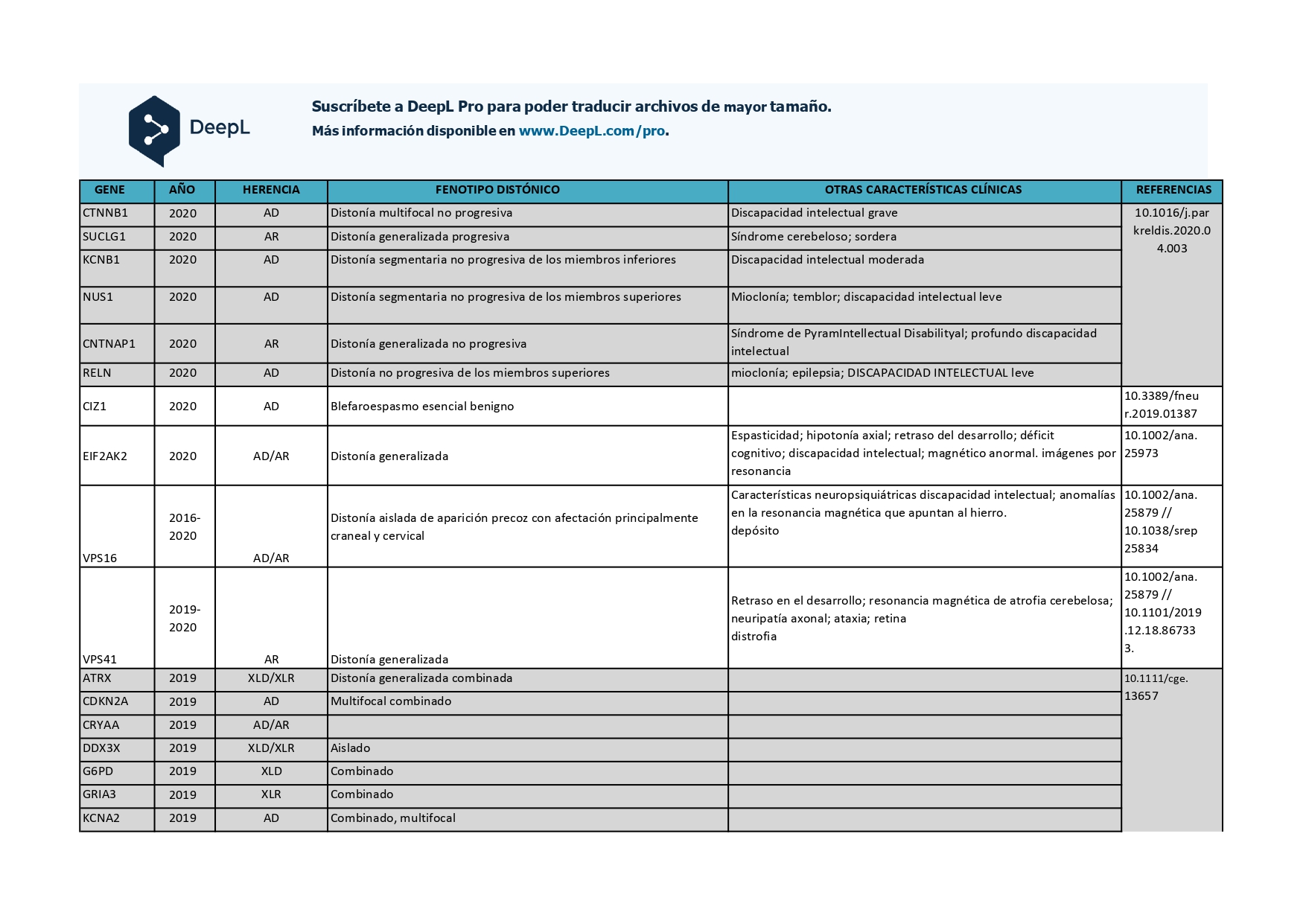
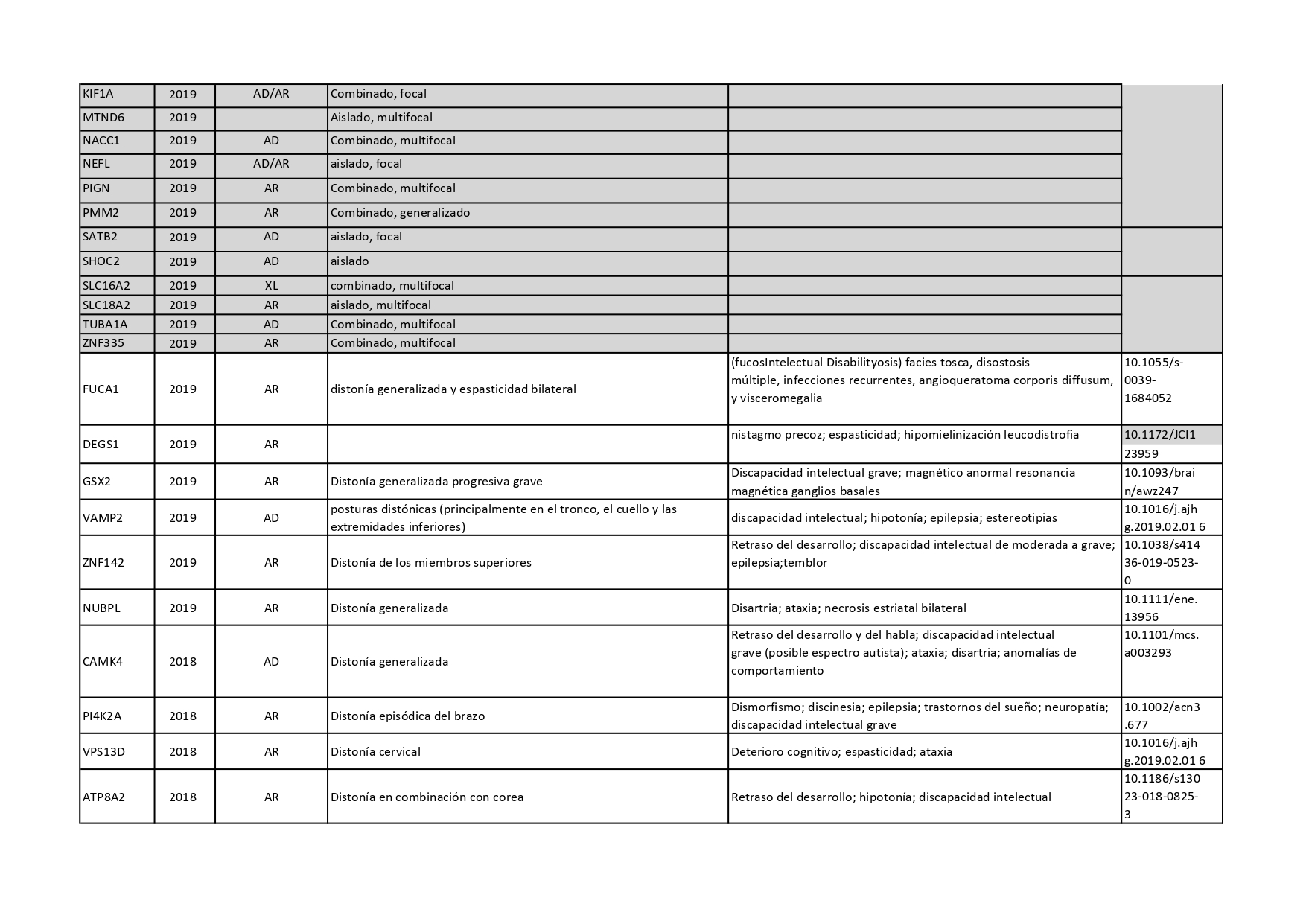
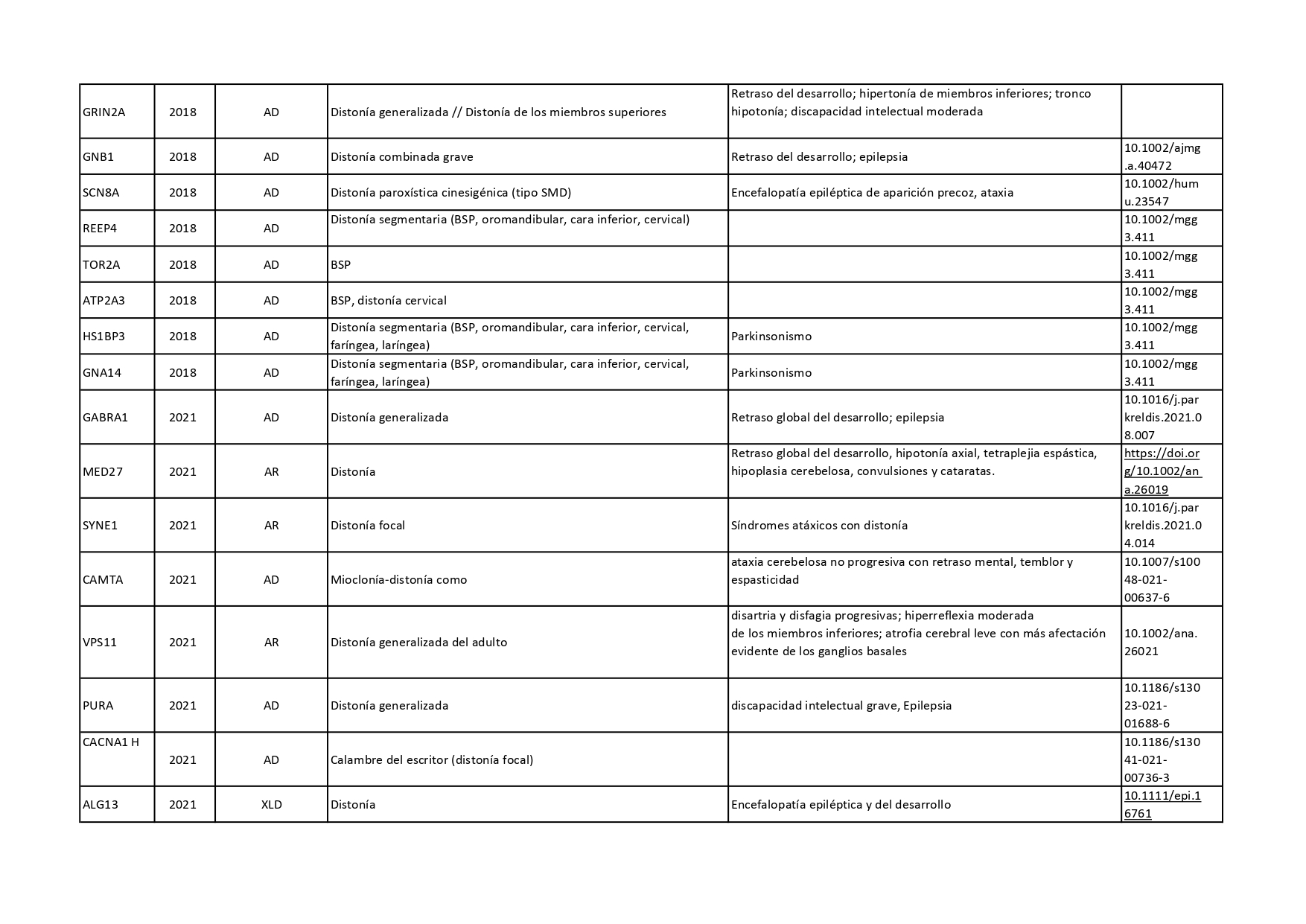
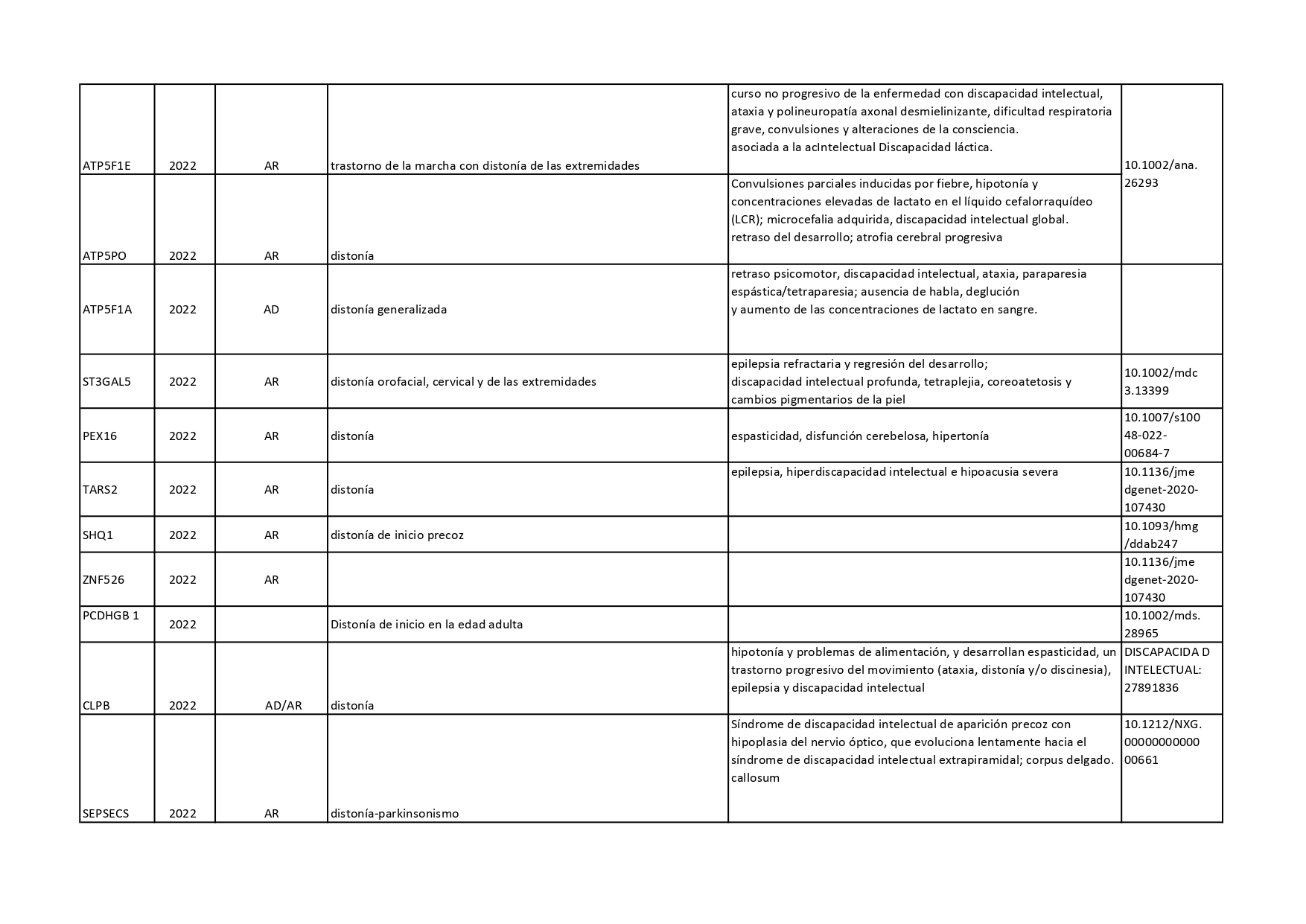
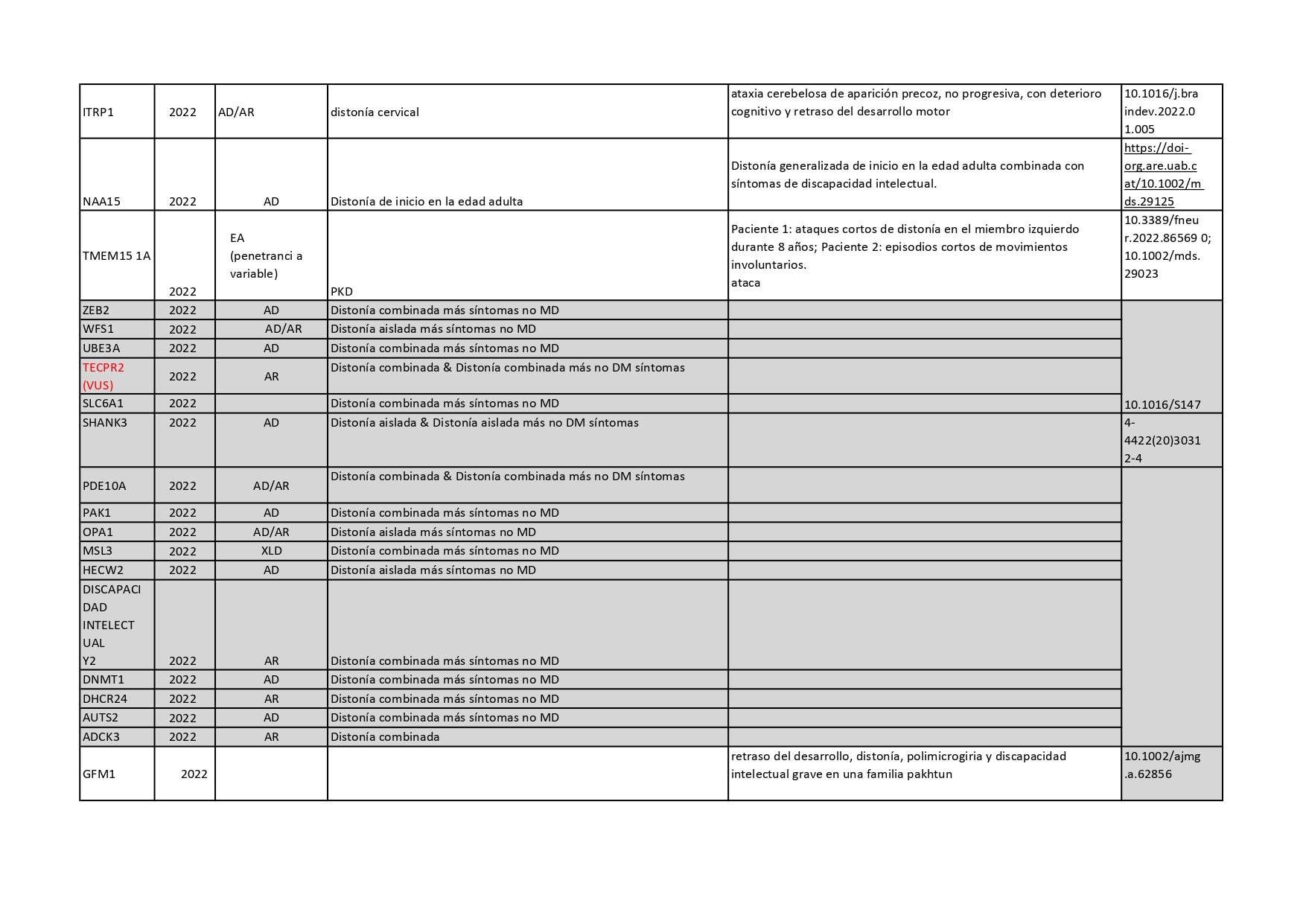
Genes que se han descrito desde 2018 hasta 2022 como asociados a distonías aisladas o en combinación con otros síntomas:
Pincha en cada diapositiva para ver el contenido
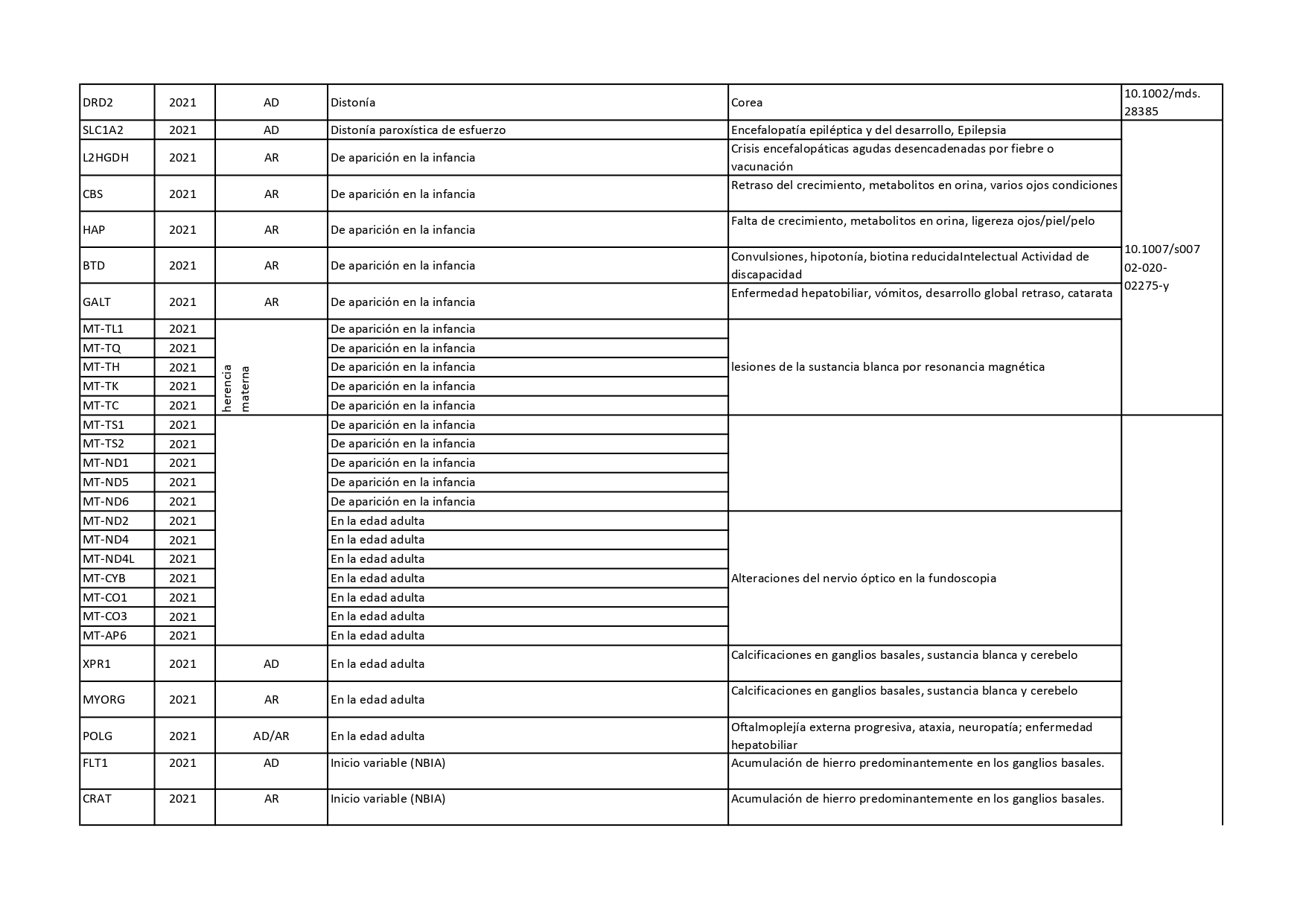
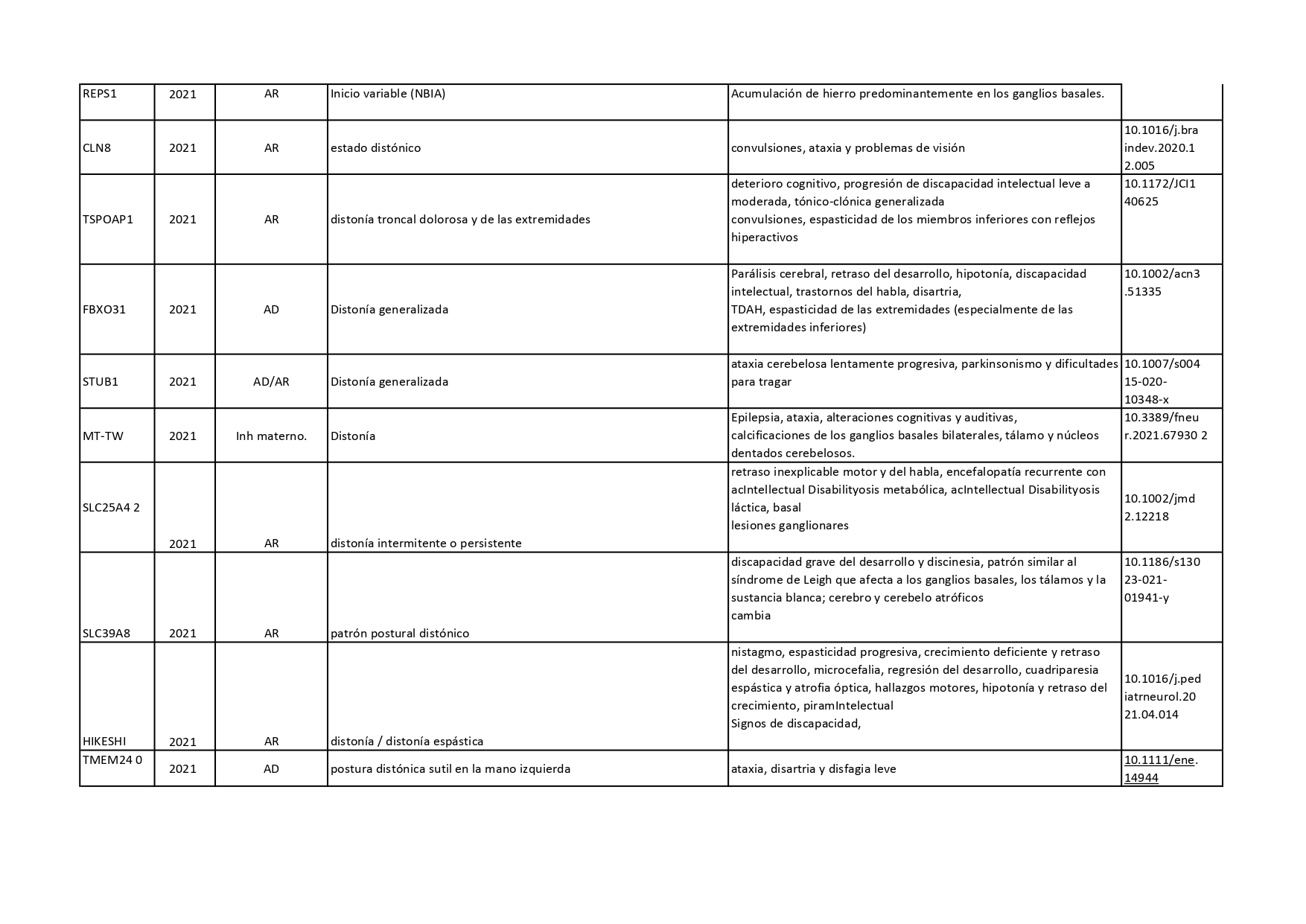
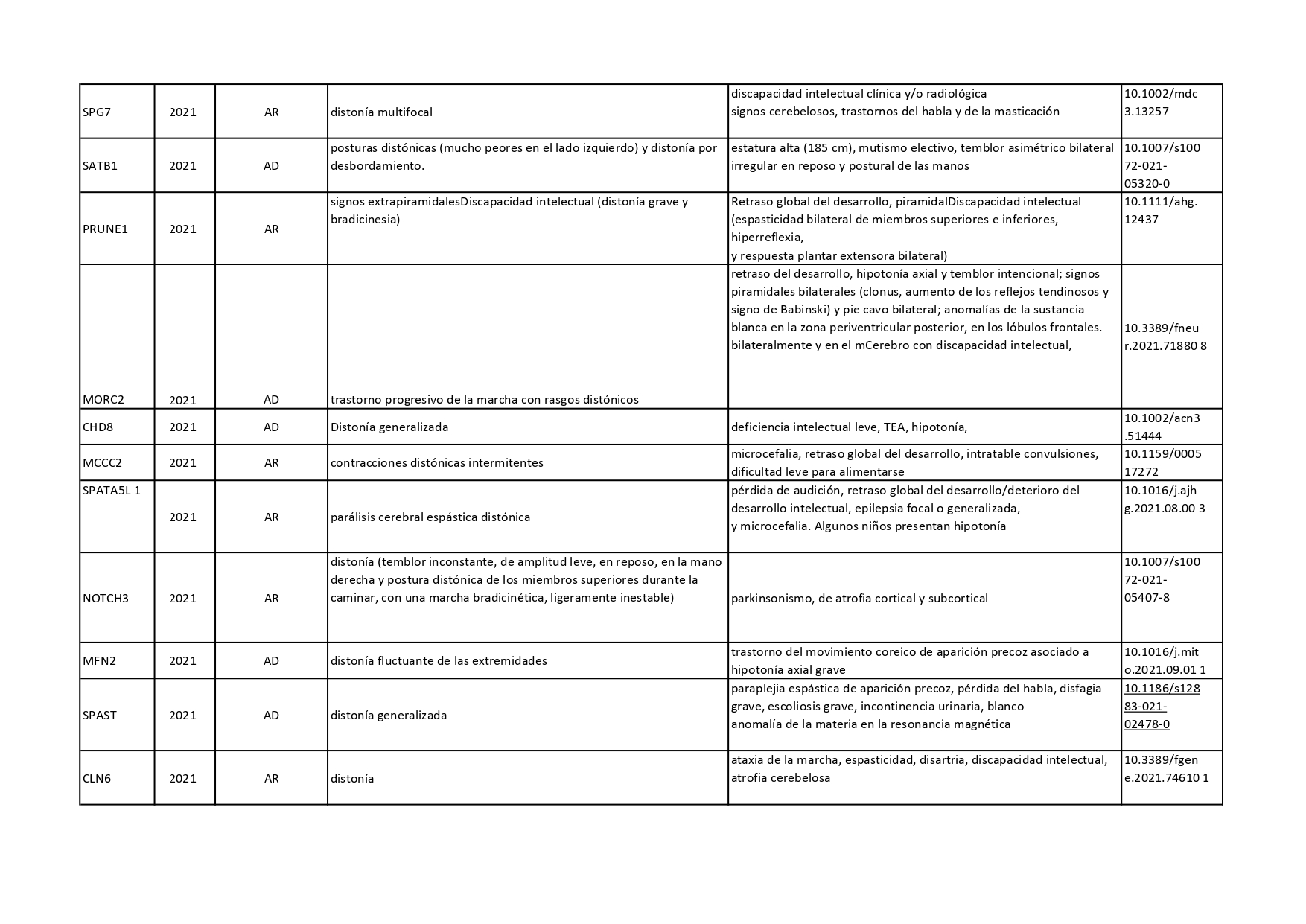
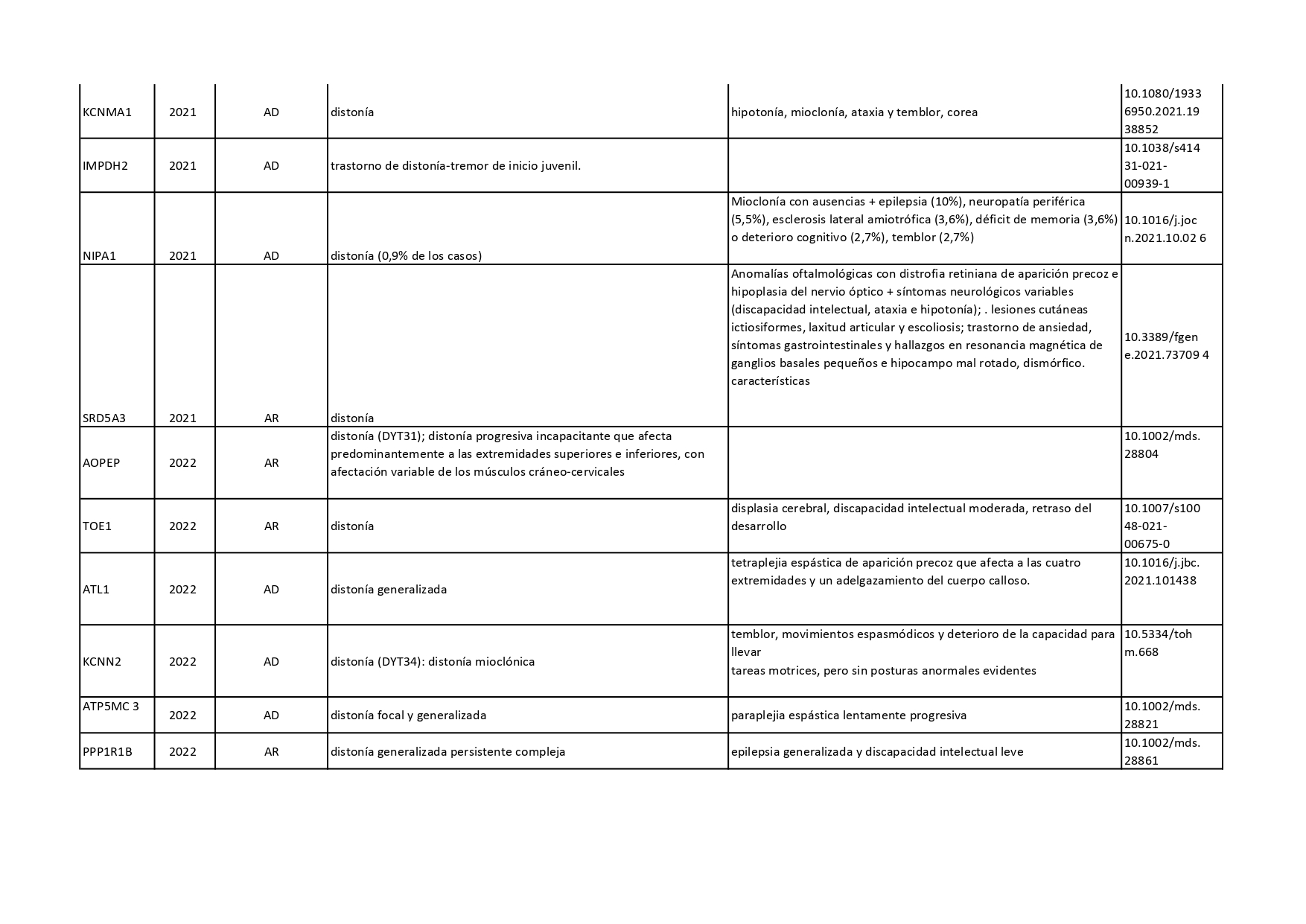
Cuando hablamos de enfermedades genéticas, debemos tener en cuenta que la alteración está presente en todas las células de nuestro organismo y no se puede eliminar. En los últimos años, se han venido desarrollando nuevas terapias génicas enfocadas a corregir el error genético de algunas enfermedades neurológicas. En el caso concreto de la distonía, se están desarrollando ensayos clínicos con una terapia génica para tratar la deficiencia de AADC (o deficiencia de L- Aminoácido aromático descarboxilasa).

En los últimos años, el estudio de las mutaciones genéticas ha ido evolucionando de manera exponencial, gracias a la aparición de técnicas mejoradas que nos permiten secuenciar nuestro genoma (el conjunto completo de instrucciones de nuestro cuerpo; todo nuestro ADN).
¿Qué es secuenciar? La secuenciación del ADN consiste en ‘leer’ y conocer la secuencia exacta que tiene cada uno de nuestros genes. Por lo tanto, según la situación, procederemos a leer nuestra biblioteca de manera diferente.
En los casos en los que existe una sospecha clínica asociada a un gen concreto, se puede proceder a analizar este gen en concreto, es decir, revisar el libro sospechoso.
Sin embargo, cuando no tenemos ninguna sospecha, debemos ir más allá y leer zonas más grandes de nuestro genoma, para lo cual tenemos varias estrategias:
